Read and Analyze 'MetIDQ™' Software Output Files.
MetAlyzer
![]()
![]()
An R Package to read and analyze MetIDQ™ output
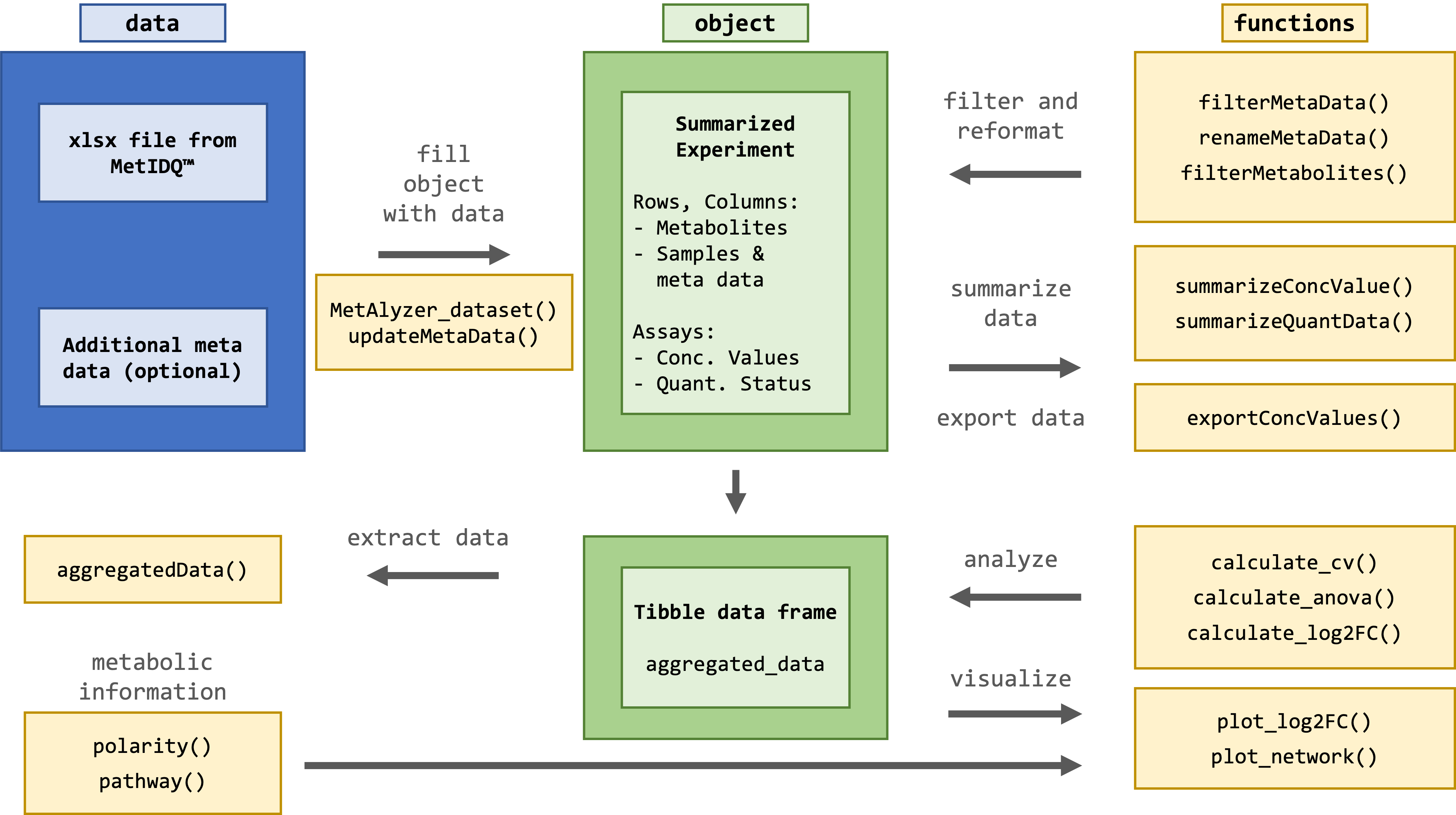
The package provides methods to read output files from the MetIDQ™ software into R. Metabolomics data is read and reformatted into an S4 object for convenient data handling, statistics and downstream analysis.
Install
There is a version available on CRAN.
install.packages("MetAlyzer")
For the latest version install from GitHub
library(devtools)
install_github("nilsmechtel/MetAlyzer")
Quickstart
The package takes metabolomic measurements and the quantification status (e.g. "Valid", "LOQ", "LOD") as ".xlsx" files generated from the MetIDQ™ software. Additionally, meta data for each sample can be provided for further analysis.
This is an extract from one of the provided example data sets. 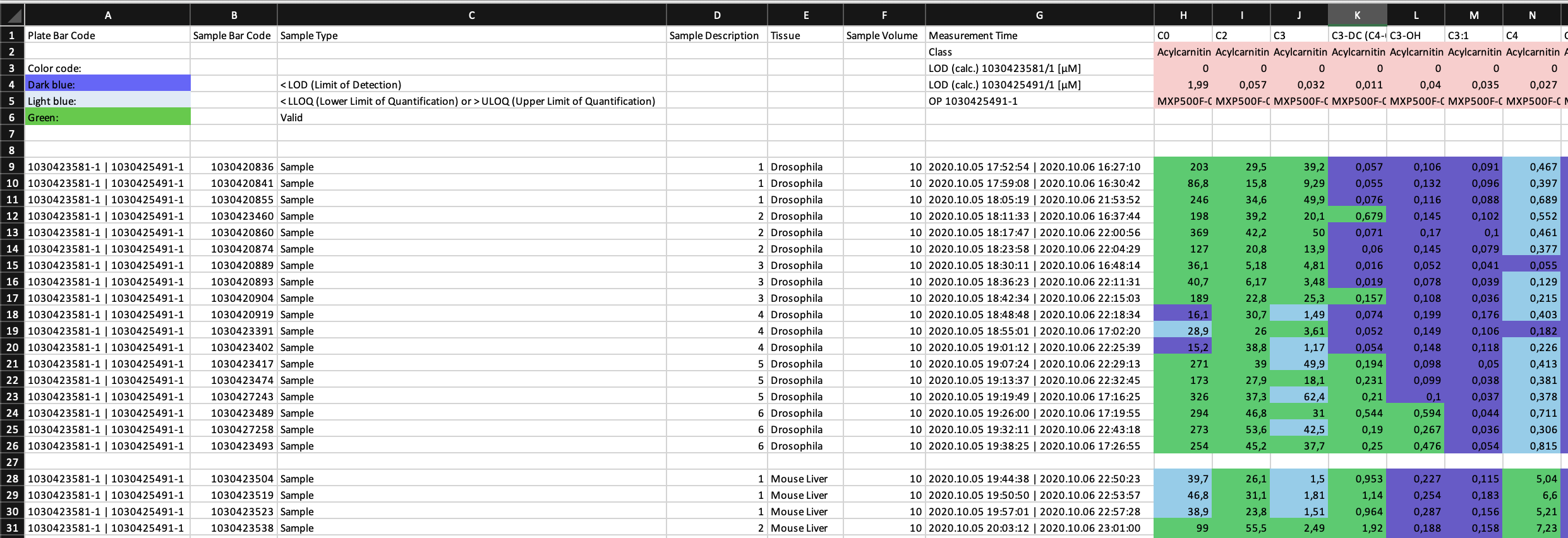
Create MetAlyzer object:
> metalyzer_se <- MetAlyzer_dataset(file_path = extraction_data())
_____ ______ _______ _________ ________ ___ ___ ___ ________ _______ ________
|\ _ \ _ \|\ ___ \|\___ ___\\ __ \|\ \ |\ \ / /|\_____ \|\ ___ \ |\ __ \
\ \ \\\__\ \ \ \ __/\|___ \ \_\ \ \|\ \ \ \ \ \ \/ / /\|___/ /\ \ __/|\ \ \|\ \
\ \ \\|__| \ \ \ \_|/__ \ \ \ \ \ __ \ \ \ \ \ / / / / /\ \ \_|/_\ \ _ _\
\ \ \ \ \ \ \ \_|\ \ \ \ \ \ \ \ \ \ \ \____ \/ / / / /_/__\ \ \_|\ \ \ \\ \|
\ \__\ \ \__\ \_______\ \ \__\ \ \__\ \__\ \_______\__/ / / |\________\ \_______\ \__\\ _\
\|__| \|__|\|_______| \|__| \|__|\|__|\|_______|\____/ / \|_______|\|_______|\|__|\|__|
\|____|/
Info: Reading color code "FFFFCCCC" as "#FFCCCC"
Info: Reading color code "FF00CD66" as "#00CD66"
Info: Reading color code "FF6A5ACD" as "#6A5ACD"
Info: Reading color code "FF87CEEB" as "#87CEEB"
Info: Reading color code "FFFFFFCC" as "#FFFFCC"
Measured concentration values:
------------------------------
0% 25% 50% 75% 100%
0.000 0.017 1.760 21.200 288149.000
NAs: 5348 (8.38%)
Note: 'Metabolism Indicators' are frequently NA!
Measured quantification status:
-------------------------------
Valid: 24095 (37.77%)
LOQ: 5799 (9.09%)
LOD: 21789 (34.16%)
Invalid: 12105 (18.98%)
NAs: 0 (0%)
Downstream analysis:
For further filtering, statistical analysis and plotting, the data is reformatted and aggregated into a tibble data frame.
> aggregatedData(metalyzer_se)
# A tibble: 63,788 × 5
# Groups: Metabolite [862]
ID Metabolite Class Concentration Status
<fct> <fct> <fct> <dbl> <fct>
1 9 C0 Acylcarnitines 203 Valid
2 10 C0 Acylcarnitines 86.8 Valid
3 11 C0 Acylcarnitines 246 Valid
4 12 C0 Acylcarnitines 198 Valid
5 13 C0 Acylcarnitines 369 Valid
6 14 C0 Acylcarnitines 127 Valid
7 15 C0 Acylcarnitines 36.1 Valid
8 16 C0 Acylcarnitines 40.7 Valid
9 17 C0 Acylcarnitines 189 Valid
10 18 C0 Acylcarnitines 16.1 LOD
# ℹ 63,778 more rows
# ℹ Use `print(n = ...)` to see more rows
Detailed instructions
For a comprehensive tutorial, please check out the MetAlyzer Vignette.