Browse and Query Clinical and Genomic Data from cBioPortal.
![]()
![]()
cbioportalR 
{cbioportalR} allows you to access cBioPortal’s genomic and clinical data sets directly through R. The package wraps cBioPortal’s API endpoints so R users can easily leverage the existing API to access genomic data on mutations, copy number alterations and fusions as well as data on tumor mutational burden (TMB), microsatellite instability status (MSI) and select clinical data points (depending on the study).
This package was created to work with both the public cBioPortal website, as well as private institutional cBioPortal instances (e.g. MSKCC, GENIE) with appropriate credentials and authentication.
This package is compatible with cBioPortal v5, but is subject to change as cBioPortal updates are released. To see if your cBioPortal instance is compatible, look for its version in the footer of the homepage or check portalVersion in the output of YOUR_CBIOPORTAL_INSTANCE/api/info. For more information on cBioPortal, see the following publications:
For full documentation on the cBioPortal API, please see the following links:
Note: If you are a MSK researcher working on IMPACT data, you should connect to MSK’s cBioPortal instance to get the most up to date IMPACT data, and you must follow the MSK-IMPACT publication guidelines when using this data
{cbioportalR} is part of the genomeverse, a collection of R packages designed to work together seamlessly to create reproducible clinico-genomic analysis pipelines.
Installation
You can install {cbioportalR} with the following code:
install.packages("cbioportalR")
Install the development version of {cbioportalR} with:
remotes::install_github("karissawhiting/cbioportalR")
Load the package:
library(cbioportalR)
Authentication
If you are using the public domain https://www.cbioportal.org/, you don’t need a token to start pulling data. If you are using a private instance of cBioPortal (like MSKCC’s institutional database), you will need to acquire a token and save it to your .Renviron file (or wherever you store credentials). Simply log in to your institution’s cBioPortal site, acquire a token (usually through the ‘Data Access Token’ link in your username menu in the upper right) and save it in your .Renviron file. This will save the token as an environmental variable so you don’t have to hard code the secret key in your scripts.
Tip: The following {usethis} function can easily find and open the .Renviron for you:
usethis::edit_r_environ()
Paste the token you were given (using the format below) in the .Renviron file and save the file changes. After saving you should restart your R session to ensure the token is saved and recognized.
CBIOPORTAL_TOKEN = 'YOUR_TOKEN'
You can test that your token was saved using:
get_cbioportal_token()
For every new R session, you need to set your database URL. The set_cbioportal_db() function will set an environmental variable for your session that tells the package which database to point to for all API calls. You can set it to point to the public database with db = 'www.cbioportal.org' or db = 'public'. If using a private database you will pass your institutions cBioPortal URL as db. This function will both set your URL and check the connection.
set_cbioportal_db(db = "public")
#> ✔ You are successfully connected!
#> ✔ base_url for this R session is now set to "www.cbioportal.org/api"
You are now set up for the remainder of your session! API calls depend on your internet connection and possibly a VPN connection so you can use the following to check your connection at any time throughout your session:
test_cbioportal_db()
#> ✔ You are successfully connected!
cBioPortal Data Model
There are many ways to identify and pull data (e.g. by study ID, by sample ID, by molecular profile ID). Having an understanding of how data is organized in cBioPortal will help you determine which functions you need. The figure below outlines the general data schema for cBioPortal and which functions access which levels of the schema:
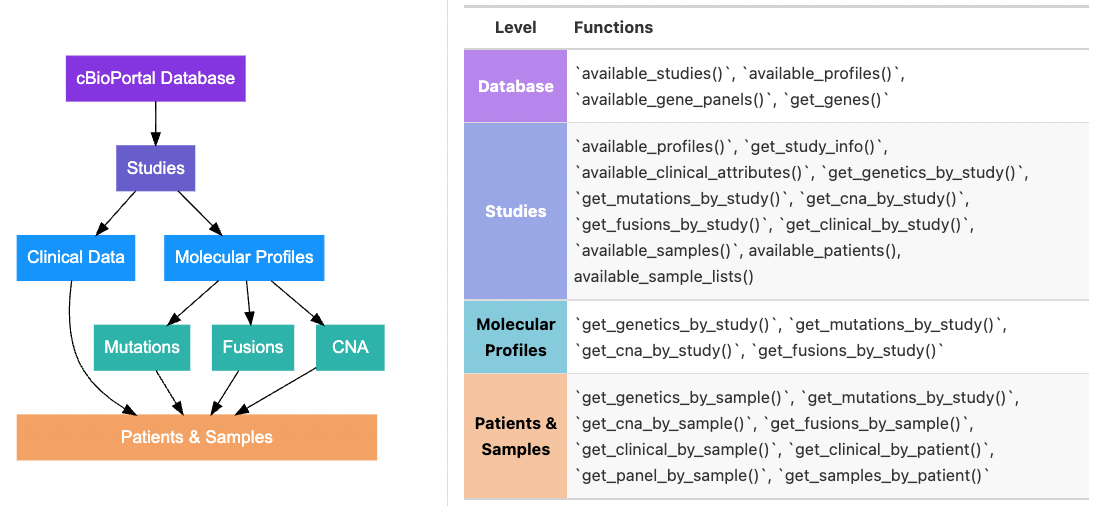
Examples
To see available studies in your database you can use:
available_studies() %>%
head(n = 10)
#> # A tibble: 10 × 13
#> studyId name description publicStudy pmid citation groups status importDate
#> <chr> <chr> <chr> <lgl> <chr> <chr> <chr> <int> <chr>
#> 1 acyc_m… Aden… Whole-exom… TRUE 2368… Ho et a… "ACYC… 0 2023-12-0…
#> 2 acyc_f… Aden… Targeted S… TRUE 2441… Ross et… "ACYC… 0 2023-12-0…
#> 3 acyc_j… Aden… Whole-geno… TRUE 2686… Rettig … "ACYC… 0 2023-12-0…
#> 4 acyc_m… Aden… WGS of 21 … TRUE 2663… Mitani … "ACYC… 0 2023-12-0…
#> 5 acyc_m… Aden… Whole-geno… TRUE 2682… Drier e… "ACYC" 0 2023-12-0…
#> 6 acyc_s… Aden… Whole exom… TRUE 2377… Stephen… "ACYC… 0 2023-12-0…
#> 7 bcc_un… Basa… Whole-exom… TRUE 2695… Bonilla… "PUBL… 0 2023-12-0…
#> 8 all_st… Acut… Comprehens… TRUE 2573… Anderss… "PUBL… 0 2023-12-0…
#> 9 ampca_… Ampu… Exome sequ… TRUE 2680… Gingras… "PUBL… 0 2023-12-0…
#> 10 all_st… Hypo… Whole geno… TRUE 2333… Holmfel… "" 0 2023-12-0…
#> # ℹ 4 more variables: allSampleCount <int>, readPermission <lgl>,
#> # cancerTypeId <chr>, referenceGenome <chr>
To view study metadata on a particular study you can use:
get_study_info("acc_tcga") %>%
t()
#> [,1]
#> name "Adrenocortical Carcinoma (TCGA, Firehose Legacy)"
#> description "TCGA Adrenocortical Carcinoma. Source data from <A HREF=\"http://gdac.broadinstitute.org/runs/stddata__2016_01_28/data/ACC/20160128/\">GDAC Firehose</A>. Previously known as TCGA Provisional."
#> publicStudy "TRUE"
#> groups "PUBLIC"
#> status "0"
#> importDate "2024-01-19 09:11:57"
#> allSampleCount "92"
#> sequencedSampleCount "90"
#> cnaSampleCount "90"
#> mrnaRnaSeqSampleCount "0"
#> mrnaRnaSeqV2SampleCount "79"
#> mrnaMicroarraySampleCount "0"
#> miRnaSampleCount "0"
#> methylationHm27SampleCount "0"
#> rppaSampleCount "46"
#> massSpectrometrySampleCount "0"
#> completeSampleCount "75"
#> readPermission "TRUE"
#> treatmentCount "0"
#> structuralVariantCount "0"
#> studyId "acc_tcga"
#> cancerTypeId "acc"
#> cancerType.name "Adrenocortical Carcinoma"
#> cancerType.dedicatedColor "Purple"
#> cancerType.shortName "ACC"
#> cancerType.parent "adrenal_gland"
#> cancerType.cancerTypeId "acc"
#> referenceGenome "hg19"
To pull all genomic data for a particular study you can use:
df <- get_genetics_by_study(study_id = "acc_tcga")
#> ℹ Returning all data for the "acc_tcga_mutations" molecular profile in the "acc_tcga" study
#> ℹ Returning all data for the "acc_tcga_gistic" molecular profile in the "acc_tcga" study
#> ! No "structural_variant" data returned. Error: No molecular profile for `data_type = fusion` found in "acc_tcga". See `available_profiles('acc_tcga')`
As a result, you will get a list of data frames with mutation and CNA data respectively. The function will also try to pull fusion (structural variant) data, but there is no fusion data available for this study, as indicated by the function message.
df$mutation %>%
head()
#> # A tibble: 6 × 28
#> hugoGeneSymbol entrezGeneId uniqueSampleKey uniquePatientKey
#> <chr> <int> <chr> <chr>
#> 1 KRT8 3856 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox…
#> 2 LCE1B 353132 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox…
#> 3 DNAH14 127602 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox…
#> 4 KLRB1 3820 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox…
#> 5 SALL2 6297 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox…
#> 6 TMEM266 123591 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox…
#> # ℹ 24 more variables: molecularProfileId <chr>, sampleId <chr>,
#> # patientId <chr>, studyId <chr>, center <chr>, mutationStatus <chr>,
#> # validationStatus <chr>, tumorAltCount <int>, tumorRefCount <int>,
#> # normalAltCount <int>, normalRefCount <int>, startPosition <int>,
#> # endPosition <int>, referenceAllele <chr>, proteinChange <chr>,
#> # mutationType <chr>, ncbiBuild <chr>, variantType <chr>, keyword <chr>,
#> # chr <chr>, variantAllele <chr>, refseqMrnaId <chr>, …
df$cna %>%
head()
#> # A tibble: 6 × 9
#> hugoGeneSymbol entrezGeneId uniqueSampleKey uniquePatientKey
#> <chr> <int> <chr> <chr>
#> 1 MEF2B 100271849 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox…
#> 2 FRS2 10818 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox…
#> 3 LRP1B 53353 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox…
#> 4 KMT2A 4297 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox…
#> 5 PC 5091 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox…
#> 6 CCNE1 898 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox…
#> # ℹ 5 more variables: molecularProfileId <chr>, sampleId <chr>,
#> # patientId <chr>, studyId <chr>, alteration <int>
You can also pull data by specific sample IDs but the API requires a bit more information from you (unlike pulling by study ID which returns everything available for that study). This can be useful when working within a very large database or working across samples housed in multiple different studies. When querying by sample_id you must also specify the corresponding study_id in which the samples are housed. When these pieces of information are not provided, {cbioportalR} makes an informed guess based on your connection and will throw an informative message to clarify exactly what is being queried. In the example below, the function defaults to the public version of the IMPACT database (study_id = "msk_impact_2017").
samples <- available_samples(study_id = "acc_tcga") %>%
pull(sampleId) %>%
head(n = 10)
mutations <- get_mutations_by_sample(sample_id = samples)
#> The following parameters were used in query:
#> Study ID: "msk_impact_2017"
#> Molecular Profile ID: "msk_impact_2017_mutations"
#> Genes: "All available genes"
# no results returned because these samples are not in this study
length(mutations) == 0
#> [1] TRUE
No results were returned because the samples are not stored in this study. When we specify the correct study (study_id = "acc_tcga"), we get accurate results. You can check which samples are stored in a study using available_samples(study_id = "acc_tcga").
mutations <- get_mutations_by_sample(sample_id = samples,
study_id = "acc_tcga")
#> The following parameters were used in query:
#> Study ID: "acc_tcga"
#> Molecular Profile ID: "acc_tcga_mutations"
#> Genes: "All available genes"
mutations %>%
head()
#> # A tibble: 6 × 28
#> hugoGeneSymbol entrezGeneId uniqueSampleKey uniquePatientKey
#> <chr> <int> <chr> <chr>
#> 1 KRT8 3856 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox…
#> 2 LCE1B 353132 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox…
#> 3 DNAH14 127602 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox…
#> 4 KLRB1 3820 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox…
#> 5 SALL2 6297 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox…
#> 6 TMEM266 123591 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox…
#> # ℹ 24 more variables: molecularProfileId <chr>, sampleId <chr>,
#> # patientId <chr>, studyId <chr>, center <chr>, mutationStatus <chr>,
#> # validationStatus <chr>, tumorAltCount <int>, tumorRefCount <int>,
#> # normalAltCount <int>, normalRefCount <int>, startPosition <int>,
#> # endPosition <int>, referenceAllele <chr>, proteinChange <chr>,
#> # mutationType <chr>, ncbiBuild <chr>, variantType <chr>, keyword <chr>,
#> # chr <chr>, variantAllele <chr>, refseqMrnaId <chr>, …
Lastly, you can also pull clinical data or sample metadata (e.g. tumor sample site) by study ID, sample ID or patient ID. To see what data is available, you can use:
available_clinical_attributes(study_id = "acc_tcga") %>%
head()
#> # A tibble: 6 × 7
#> displayName description datatype patientAttribute priority clinicalAttributeId
#> <chr> <chr> <chr> <lgl> <chr> <chr>
#> 1 Diagnosis … Age at whi… NUMBER TRUE 1 AGE
#> 2 Neoplasm D… The extent… STRING TRUE 1 AJCC_PATHOLOGIC_TU…
#> 3 American J… The versio… STRING TRUE 1 AJCC_STAGING_EDITI…
#> 4 Atypical M… Atypical M… STRING TRUE 1 ATYPICAL_MITOTIC_F…
#> 5 Cancer Type Cancer type STRING FALSE 1 CANCER_TYPE
#> 6 Cancer Typ… Cancer typ… STRING FALSE 1 CANCER_TYPE_DETAIL…
#> # ℹ 1 more variable: studyId <chr>
get_clinical_by_study("acc_tcga")
#> ! Sample Level Clinical Data: No `clinical_attribute` passed. Defaulting to returning all clinical attributes in "acc_tcga" study
#> ! Patient Level Clinical Data: No `clinical_attribute` passed. Defaulting to returning all clinical attributes in "acc_tcga" study
#> # A tibble: 6,292 × 7
#> uniquePatientKey patientId studyId clinicalAttributeId value dataLevel
#> <chr> <chr> <chr> <chr> <chr> <chr>
#> 1 VENHQS1PUi1BNUoxOmFjY1… TCGA-OR-… acc_tc… AGE 58 PATIENT
#> 2 VENHQS1PUi1BNUoxOmFjY1… TCGA-OR-… acc_tc… AJCC_PATHOLOGIC_TU… Stag… PATIENT
#> 3 VENHQS1PUi1BNUoxOmFjY1… TCGA-OR-… acc_tc… ATYPICAL_MITOTIC_F… Atyp… PATIENT
#> 4 VENHQS1PUi1BNUoxOmFjY1… TCGA-OR-… acc_tc… CAPSULAR_INVASION Inva… PATIENT
#> 5 VENHQS1PUi1BNUoxOmFjY1… TCGA-OR-… acc_tc… CLIN_M_STAGE M0 PATIENT
#> 6 VENHQS1PUi1BNUoxOmFjY1… TCGA-OR-… acc_tc… CT_SCAN_PREOP_RESU… [Unk… PATIENT
#> 7 VENHQS1PUi1BNUoxOmFjY1… TCGA-OR-… acc_tc… CYTOPLASM_PRESENCE… Cyto… PATIENT
#> 8 VENHQS1PUi1BNUoxOmFjY1… TCGA-OR-… acc_tc… DAYS_TO_INITIAL_PA… 0 PATIENT
#> 9 VENHQS1PUi1BNUoxOmFjY1… TCGA-OR-… acc_tc… DFS_MONTHS 24.77 PATIENT
#> 10 VENHQS1PUi1BNUoxOmFjY1… TCGA-OR-… acc_tc… DFS_STATUS 1:Re… PATIENT
#> # ℹ 6,282 more rows
#> # ℹ 1 more variable: sampleId <chr>
get_clinical_by_sample(sample_id = samples, study_id = "acc_tcga") %>%
head(10)
#> ! No `clinical_attribute` passed. Defaulting to returning
#> all clinical attributes in "acc_tcga" study
#> # A tibble: 10 × 7
#> uniqueSampleKey uniquePatientKey sampleId patientId studyId
#> <chr> <chr> <chr> <chr> <chr>
#> 1 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox… TCGA-OR… TCGA-OR-… acc_tc…
#> 2 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox… TCGA-OR… TCGA-OR-… acc_tc…
#> 3 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox… TCGA-OR… TCGA-OR-… acc_tc…
#> 4 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox… TCGA-OR… TCGA-OR-… acc_tc…
#> 5 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox… TCGA-OR… TCGA-OR-… acc_tc…
#> 6 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox… TCGA-OR… TCGA-OR-… acc_tc…
#> 7 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox… TCGA-OR… TCGA-OR-… acc_tc…
#> 8 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox… TCGA-OR… TCGA-OR-… acc_tc…
#> 9 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox… TCGA-OR… TCGA-OR-… acc_tc…
#> 10 VENHQS1PUi1BNUoxLTAxOmFjY190Y2dh VENHQS1PUi1BNUox… TCGA-OR… TCGA-OR-… acc_tc…
#> # ℹ 2 more variables: clinicalAttributeId <chr>, value <chr>
patients <- available_patients(study_id = "acc_tcga") %>%
pull(patientId) %>%
head(n = 10)
get_clinical_by_patient(patient_id = patients, study_id = "acc_tcga",
clinical_attribute = "AGE") %>%
head(10)
All functions that pull by study IDs are limited to pulling data from one study at a time. If you need to pull specific samples from multiple studies, you likely want to pull by sample ID (instead of study ID) and supply the function with a dataframe of sample_study_pairs that specify where the function should look for each study. For more information see the Overview of Workflow Vignette.
Contributing
Please note that {cbioportalR} is released with a Contributor Code of Conduct. By contributing to this project, you agree to abide by its terms.
Thank you to contributors!
@arorarshi, @AxelitoMartin, @edrill, @jalavery, @ddsjoberg@karomanchuk@hfuchs5@alrein-05@stl2137
Thank you Isaak Liptzin for the hex sticker!