Create HIDECAN Plots for Visualising Genome-Wide Association Studies and Differential Expression R….
hidecan
hidecan is an R package for generating HIDECAN plots, which are visualisations summarising the results of one or more Genome-wide association study (GWAS) and transcriptomics differential expression (DE) analysis, alongside candidate genes of interest.
Installation
You can install the development version of hidecan from GitHub with:
# install.packages("devtools")
devtools::install_github("PlantandFoodResearch/hidecan")
Usage
The hidecan package works as follows:
it takes as an input one of more data-frames containing GWAS results, differential expression results and list of candidate genes of interest;
it computes the length of each chromosome based on the genomic position of the markers and genes provided in the input data;
it filters the datasets to retain significant markers or differentially expressed genes, according to a threshold on their score and/or log2-fold change. The fold-change is set by the user, and can be different for GWAS and differential expression results.
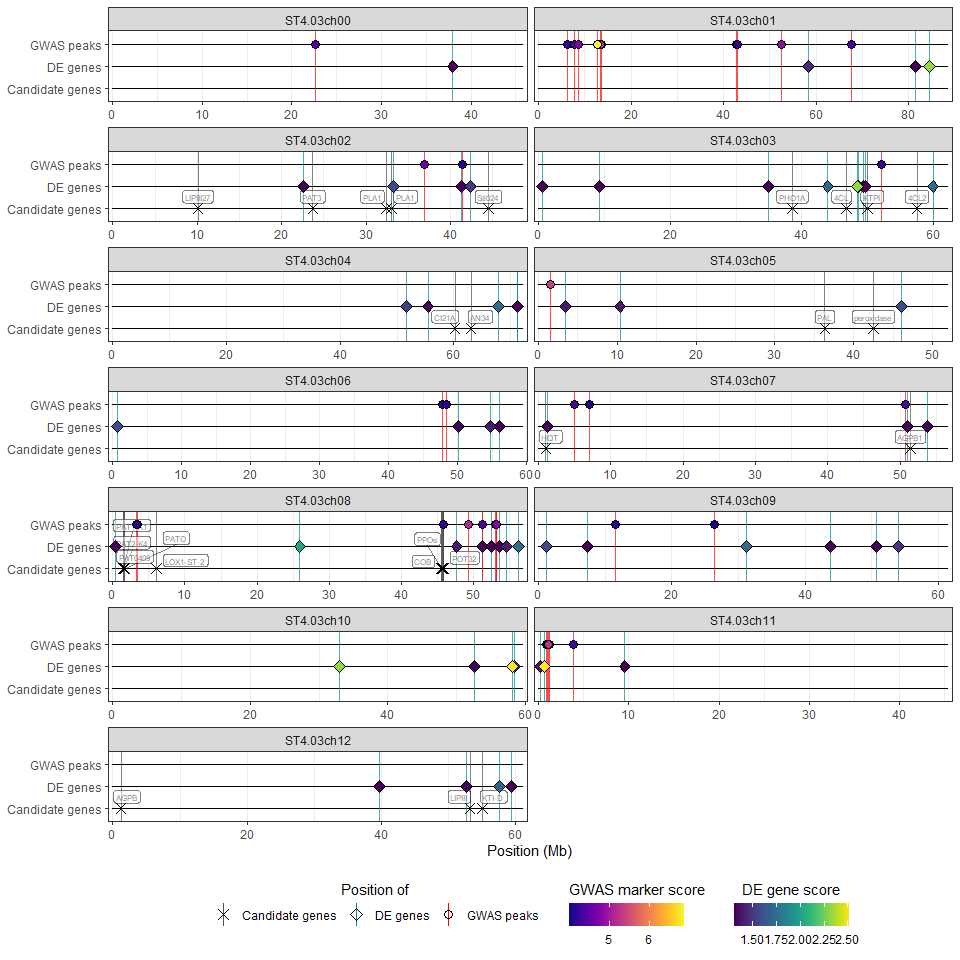
it displays the position of the significant markers and genes alongside candidate genes (HIDECAN plot). The plot can be customised by the user via a number of parameters (e.g. legend position or label size).
The wrapper function hidecan_plot() performs all of these steps. Its use is demonstrated below with an example dataset:
library(hidecan)
## Getting an example dataset
x <- get_example_data()
hidecan_plot(
gwas_list = x[["GWAS"]], ## data-frame of GWAS results
de_list = x[["DE"]], ## data-frame of DE results
can_list = x[["CAN"]], ## data-frame of candidate genes
score_thr_gwas = -log10(0.0001), ## sign. threshold for GWAS
score_thr_de = -log10(0.05), ## sign. threshold for DE
log2fc_thr = 0, ## log2FC threshold for DE
label_size = 2 ## label size for candidate genes
)