Tools for Regression Using Pre-Computed Summary Statistics.
pcsstools 
![]()
Overview
pcsstools is an R package to describe various regression models using only pre-computed summary statistics (PCSS) from genome-wide association studies (GWASs) and PCSS repositories such as GeneAtlas. This eliminates the logistic, privacy, and access concerns that accompany the use of individual patient-level data (IPD).
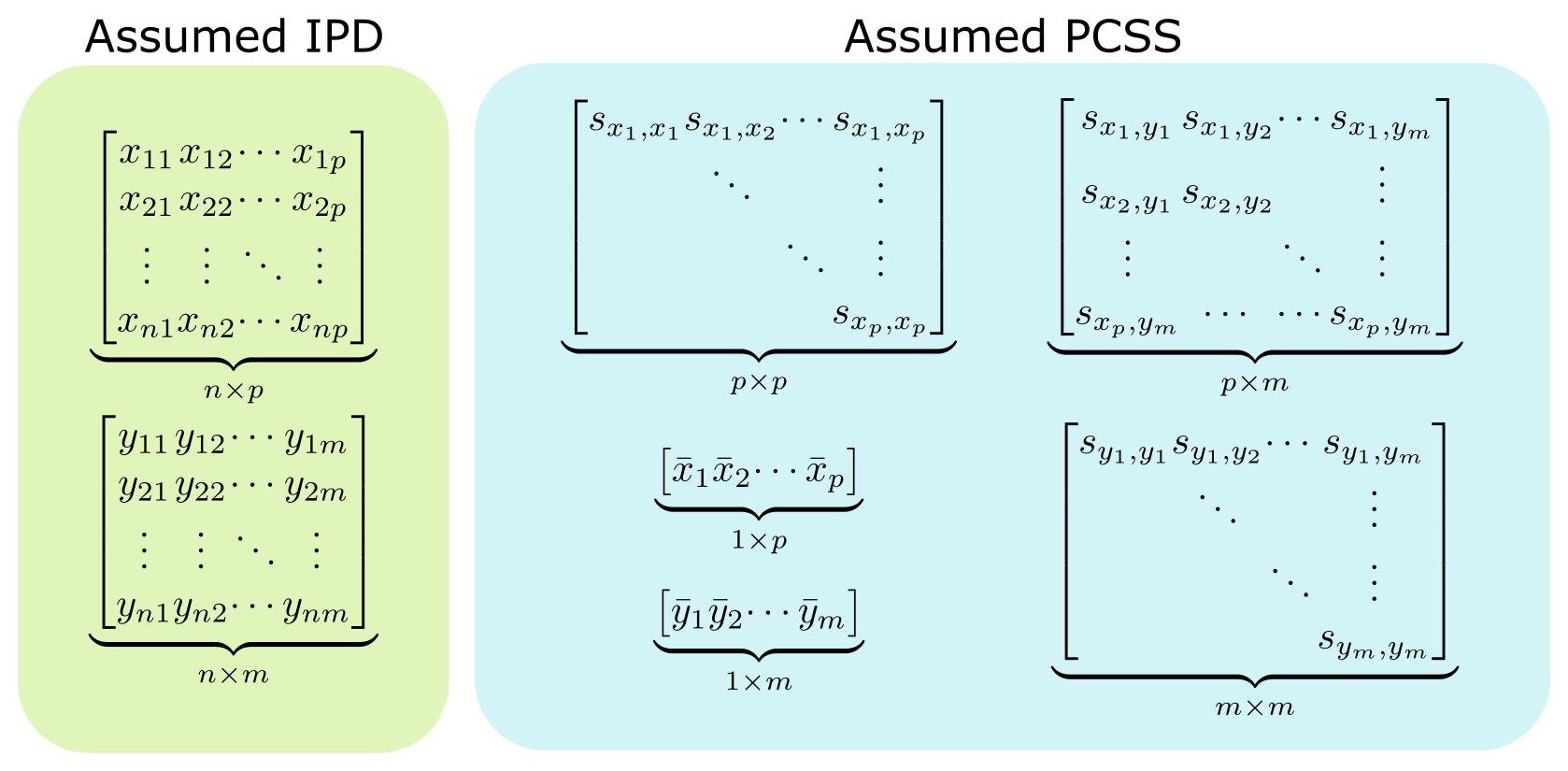
The following figure highlights the information typically needed to perform regression analysis on a set of m phenotypes with p covariates when IPD is available, and the PCSS that are commonly needed to approximate this same model in pcsstools.
Currently, pcsstools supports the linear modeling of complex phenotypes defined via functions of other phenotypes. Supported functions include:
- linear combinations (e.g. ϕ1y1 + ϕ2y2)
- products (e.g. y1 ∘ y2)
- logical combinations (e.g. y1 ∧ y2 or y1 ∨ y2)
Installation
You can install pcsstools from CRAN with
install.packages("pcsstools")
Development Version
You can install the in-development version of pcsstools from GitHub with
# install.packages("devtools")
devtools::install_github("jackmwolf/pcsstools")
Examples
We will walk through two examples using pcsstools to model combinations of phenotypes using PCSS and then compare our results to those found using IPD.
library(pcsstools)
Principal Component Analysis
Let’s model the first principal component score of three phenotypes using PCSS.
First, we’ll load in some data. We have three SNPs; minor allele counts (g1, g2, and g3), a continuous covariate (x1), and three continuous phenotypes (y1, y2, and y3).
dat <- pcsstools_example[c("g1", "g2", "g3", "x1", "y1", "y2", "y3")]
head(dat)
#> g1 g2 g3 x1 y1 y2 y3
#> 1 0 1 1 0.04239463 -0.1416907 1.19902689 -1.10982855
#> 2 1 0 0 1.35306987 0.6822496 -1.19624311 -0.97103574
#> 3 0 1 0 -1.01226388 0.8337136 0.75777722 -1.02609693
#> 4 0 1 0 -0.35358877 -0.1718187 1.13433957 -0.08290115
#> 5 1 1 0 1.20242824 0.5528258 -0.07515538 -2.43725278
#> 6 0 1 0 0.20310211 -0.9358902 -0.75434908 -1.59552034
First, we need our assumed summary statistics: means, the full covariance matrix, and our sample size.
pcss <- list(
means = colMeans(dat),
covs = cov(dat),
n = nrow(dat)
)
Then, we can calculate the linear model by using pcsslm(). Our formula will list all phenotypes as one sum, joined together by + operators and we indicate that we want the first principal component score by setting comp = 1. We also want to center and standardize y1, y2, and y3 before computing principal component scores; we will do so by setting center = TRUE and standardize = TRUE.
model_pcss <- pcsslm(y1 + y2 + y3 ~ g1 + g2 + g3 + x1, pcss = pcss, comp = 1,
center = TRUE, standardize = TRUE)
model_pcss
#> Model approximated using Pre-Computed Summary Statistics.
#>
#> Call:
#> pcsslm(formula = y1 + y2 + y3 ~ g1 + g2 + g3 + x1, pcss = pcss,
#> comp = 1, center = TRUE, standardize = TRUE)
#>
#> Coefficients:
#> Estimate Std. Error t value Pr(>|t|)
#> (Intercept) -0.03166 0.04710 -0.672 0.501581
#> g1 0.30333 0.04264 7.115 2.14e-12 ***
#> g2 -0.10125 0.03725 -2.718 0.006681 **
#> g3 -0.20014 0.05391 -3.713 0.000216 ***
#> x1 0.96190 0.02603 36.953 < 2e-16 ***
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#>
#> Residual standard error: 0.8234 on 995 degrees of freedom
#> Multiple R-squared: 0.5891, Adjusted R-squared: 0.5874
#> F-statistic: 356.6 on 4 and 995 DF, p-value: < 2.2e-16
Here’s the same model using individual patient data.
pc_1 <- prcomp(x = dat[c("y1", "y2", "y3")], center = TRUE, scale. = TRUE)$x[, "PC1"]
model_ipd <- lm(pc_1 ~ g1 + g2 + g3 + x1, data = dat)
summary(model_ipd)
#>
#> Call:
#> lm(formula = pc_1 ~ g1 + g2 + g3 + x1, data = dat)
#>
#> Residuals:
#> Min 1Q Median 3Q Max
#> -2.48100 -0.55738 -0.00702 0.56556 2.42936
#>
#> Coefficients:
#> Estimate Std. Error t value Pr(>|t|)
#> (Intercept) 0.03166 0.04710 0.672 0.501581
#> g1 -0.30333 0.04264 -7.115 2.14e-12 ***
#> g2 0.10125 0.03725 2.718 0.006681 **
#> g3 0.20014 0.05391 3.713 0.000216 ***
#> x1 -0.96190 0.02603 -36.953 < 2e-16 ***
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#>
#> Residual standard error: 0.8234 on 995 degrees of freedom
#> Multiple R-squared: 0.5891, Adjusted R-squared: 0.5874
#> F-statistic: 356.6 on 4 and 995 DF, p-value: < 2.2e-16
In this case, our coefficient estimates are off by a factor of -1; this is because we picked the opposite vector of principal component weights to prcomp. This distinction in sign is arbitrary (see the note in ?prcomp).
We can also compare this model to a smaller model using anova and find the same results when using both PCSS and IPD.
model_pcss_reduced <- update(model_pcss, . ~ . - g1 - g2 - g3)
anova(model_pcss_reduced, model_pcss)
#> Analysis of Variance Table
#>
#> Model 1: y1 + y2 + y3 ~ x1
#> Model 2: y1 + y2 + y3 ~ g1 + g2 + g3 + x1
#> Res.Df RSS Df Sum of Sq F Pr(>F)
#> 1 998 723.16
#> 2 995 674.60 3 48.564 23.877 6.322e-15 ***
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
model_ipd_reduced <-update(model_ipd, . ~ . - g1 - g2 - g3)
anova(model_ipd_reduced, model_ipd)
#> Analysis of Variance Table
#>
#> Model 1: pc_1 ~ x1
#> Model 2: pc_1 ~ g1 + g2 + g3 + x1
#> Res.Df RSS Df Sum of Sq F Pr(>F)
#> 1 998 723.16
#> 2 995 674.60 3 48.564 23.877 6.322e-15 ***
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Logical Combination
In this example we will approximate a linear model where our response is the logical combination “y4 or y5” (y4 ∨ y5).
First we need data with binary phenotypes.
dat <- pcsstools_example[c("g1", "g2", "x1", "y4", "y5")]
head(dat)
#> g1 g2 x1 y4 y5
#> 1 0 1 0.04239463 1 0
#> 2 1 0 1.35306987 0 1
#> 3 0 1 -1.01226388 1 1
#> 4 0 1 -0.35358877 1 0
#> 5 1 1 1.20242824 0 0
#> 6 0 1 0.20310211 1 1
Once again we will organized our assumed PCSS. In addition to the summary statistics we needed for the previous example, we also need to describe the distributions of both of our predictors through objects of class predictor. (See ?new_predictor.) pcsstools has shortcut functions to create predictor objects for common types of variables, which we will use to create a list of predictors.
pcss <- list(
means = colMeans(dat),
covs = cov(dat),
n = nrow(dat),
predictors = list(
g1 = new_predictor_snp(maf = mean(dat$g1) / 2),
g2 = new_predictor_snp(maf = mean(dat$g2) / 2),
x1 = new_predictor_normal(mean = mean(dat$x1), sd = sd(dat$x1))
)
)
class(pcss$predictors[[1]])
#> [1] "predictor"
Then we can approximate the linear model using pcsslm().
model_pcss <- pcsslm(y4 | y5 ~ g1 + g2 + x1, pcss = pcss)
model_pcss
#> Model approximated using Pre-Computed Summary Statistics.
#>
#> Call:
#> pcsslm(formula = y4 | y5 ~ g1 + g2 + x1, pcss = pcss)
#>
#> Coefficients:
#> Estimate Std. Error t value Pr(>|t|)
#> (Intercept) 0.75383 0.01995 37.786 < 2e-16 ***
#> g1 -0.05257 0.01904 -2.761 0.00587 **
#> g2 0.11709 0.01664 7.038 3.62e-12 ***
#> x1 -0.08160 0.01163 -7.019 4.14e-12 ***
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#>
#> Residual standard error: 0.3678 on 996 degrees of freedom
#> Multiple R-squared: 0.09521, Adjusted R-squared: 0.09249
#> F-statistic: 34.94 on 3 and 996 DF, p-value: < 2.2e-16
And here’s the result we would get using IPD:
model_ipd <- lm(y4 | y5 ~ g1 + g2 + x1, data = dat)
summary(model_ipd)
#>
#> Call:
#> lm(formula = y4 | y5 ~ g1 + g2 + x1, data = dat)
#>
#> Residuals:
#> Min 1Q Median 3Q Max
#> -0.97250 -0.02297 0.12654 0.22802 0.54225
#>
#> Coefficients:
#> Estimate Std. Error t value Pr(>|t|)
#> (Intercept) 0.74555 0.01970 37.849 < 2e-16 ***
#> g1 -0.06695 0.01880 -3.561 0.000387 ***
#> g2 0.13714 0.01643 8.349 2.28e-16 ***
#> x1 -0.08372 0.01148 -7.293 6.16e-13 ***
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#>
#> Residual standard error: 0.3631 on 996 degrees of freedom
#> Multiple R-squared: 0.1179, Adjusted R-squared: 0.1153
#> F-statistic: 44.39 on 3 and 996 DF, p-value: < 2.2e-16
Future Work
Support function notation for linear combinations of phenotypes (e.g.
y1 - y2 + 0.5 * y3 ~ 1 + g + x) instead of requiring a separate vector of weightsSupport functions using
.and-in the dependent variable (e.g.y1 ~ .,y1 ~ . -x)Write a vignette
References
Following are the key references for the functions in this package
Wolf, J.M., Westra, J., and Tintle, N. (2021). Using summary statistics to model multiplicative combinations of initially analyzed phenotypes with a flexible choice of covariates. Frontiers in Genetics, 25, 1962. https://doi.org/10.3389/fgene.2021.745901.
Wolf, J.M., Barnard, M., Xueting, X., Ryder, N., Westra, J., and Tintle, N. (2020). Computationally efficient, exact, covariate-adjusted genetic principal component analysis by leveraging individual marker summary statistics from large biobanks. Pacific Symposium on Biocomputing, 25, 719-730. https://doi.org/10.1142/9789811215636_0063.
Gasdaska A., Friend D., Chen R., Westra J., Zawistowski M., Lindsey W. and Tintle N. (2019) Leveraging summary statistics to make inferences about complex phenotypes in large biobanks. Pacific Symposium on Biocomputing, 24, 391-402. https://doi.org/10.1142/9789813279827_0036.