Mutation Models for Pedigree Likelihood Computations.
pedmut
Introduction
The pedmut package is part of the ped suite ecosystem for pedigree analysis in R. Its aim is to provide a framework for modelling mutations in pedigree computations.
Although pedmut is self-contained, its main purpose is to be imported by other ped suite packages, like pedprobr (marker probabilities and pedigree likelihoods), forrel (forensic pedigree analysis) and dvir.
For the theoretical background of mutation models and their properties (stationarity, reversibility, lumpability), I recommend Chapter 5 of Pedigree analysis in R, and the references therein.
Installation
# The easiest way to get `pedmut` is to install the entire `ped suite`:
install.packages("pedsuite")
# Alternatively, you can install just `pedmut`:
install.packages("pedmut")
# If you need the latest development version, install it from GitHub:
# install.packages("devtools")
devtools::install_github("magnusdv/pedmut")
A simple likelihood example
The examples below require the packages pedtools and pedprobr in addition to pedmut. The first two are core members of the ped suite and can be loaded collectively with library(pedsuite).
library(pedsuite)
library(pedmut)
The figure below shows a father and son who are homozygous for different alleles. We assume that the locus is an autosomal marker with two alleles, labelled 1 and 2.
# Create pedigree
x = nuclearPed(father = "fa", mother = "mo", child = "boy")
# Add marker
x = addMarker(x, fa = "1/1", boy = "2/2")
# Plot with genotypes
plot(x, marker = 1)
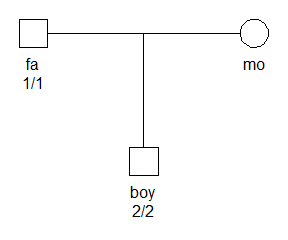
The data clearly constitutes a Mendelian error, and gives a likelihood of 0 without mutation modelling:
likelihood(x, marker = 1)
#> [1] 0
The following code sets a simple mutation model and recomputes the pedigree likelihood.
x2 = setMutationModel(x, marker = 1, model = "equal", rate = 0.1)
likelihood(x2, marker = 1)
#> [1] 0.0125
Under the mutation model, the combination of genotypes is no longer impossible, yielding a non-zero likelihood. To see details about the mutation model, we can use the mutmod() accessor:
mutmod(x2, marker = 1)
#> Unisex mutation matrix:
#> 1 2
#> 1 0.9 0.1
#> 2 0.1 0.9
#>
#> Model: equal
#> Rate: 0.1
#> Frequencies: 0.5, 0.5
#>
#> Stationary: Yes
#> Reversible: Yes
#> Lumpable: Always
Mutation models
A mutation matrix in pedmut is a stochastic matrix, with each row summing to 1, where the rows and columns are named with allele labels.
Two central functions of package are mutationMatrix() and mutationModel(). The first constructs a single mutation matrix according to various model specifications. The second produces what is typically required in applications, namely a list of two mutation matrices, named “male” and “female”.
The mutation models currently implemented in pedmut are:
equal: All mutations equally likely; probability1-rateof no mutation. Parameters:rate.proportional: Mutation probabilities are proportional to the target allele frequencies. Parameters:rate,afreq.random: This produces a matrix of random numbers, each row normalised to have sum 1. Parameters:seed.custom: Allows any valid mutation matrix to be provided by the user. Parameters:matrix.onestep: Applicable if all alleles are integers. Mutations are allowed only to the nearest integer neighbour. Parameters:rate.stepwise: For this model alleles must be integers or decimal numbers with a single decimal, such as ‘17.1’, indicating a microvariant. Mutation rates depend on whether transitions are within the same group or not, i.e., between integer alleles and microvariants in the latter case. Mutations also depend on the size of the mutation as modelled by the parameterrange, the relative probability of mutating n+1 steps versus mutating n steps. Parameters:rate,rate2,range.trivial: Diagonal mutation matrix with 1 on the diagonal. Parameters: None.
Model properties
Several properties of mutation models are of interest (both theoretical and practical) for likelihood computations. The pedmut package provides utility functions for quickly checking these:
isStationary(M, afreq): Checks ifafreqis a right eigenvector of the mutation matrixM. Stationary models have the desirable property that allele frequencies don’t change across generations.isReversible(M, afreq): Checks ifMtogether withafreqform a reversible Markov chain, i.e., that they satisfy the detailed balance criterion.isLumpable(M, lump): Checks ifMallows clustering (“lumping”) of a given subset of alleles. This implements the necessary and sufficient condition of strong lumpability of Kemeny and Snell (Finite Markov Chains, 1976).alwaysLumpable(M): Checks ifMallows lumping of any allele subset.
Further examples
An equal model with rate 0.1:
mutationMatrix("equal", rate = 0.1, alleles = c("a", "b", "c"))
#> a b c
#> a 0.90 0.05 0.05
#> b 0.05 0.90 0.05
#> c 0.05 0.05 0.90
#>
#> Model: equal
#> Rate: 0.1
#>
#> Lumpable: Always
Next, a proportional model with rate 0.1. Note that this model depends on the allele frequencies.
mutationMatrix("prop", rate = 0.1, alleles = c("a", "b", "c"), afreq = c(0.7, 0.2, 0.1))
#> a b c
#> a 0.93478261 0.04347826 0.02173913
#> b 0.15217391 0.82608696 0.02173913
#> c 0.15217391 0.04347826 0.80434783
#>
#> Model: proportional
#> Rate: 0.1
#> Frequencies: 0.7, 0.2, 0.1
#>
#> Stationary: Yes
#> Reversible: Yes
#> Lumpable: Always
To illustrate the stepwise model, we recreate the mutation matrix in Section 2.1.3 of Simonsson and Mostad (FSI:Genetics, 2015). This is done as follows:
mutationMatrix(model = "stepwise", alleles = c("16", "17", "18", "16.1", "17.1"),
rate = 0.003, rate2 = 0.001, range = 0.5)
#> 16 17 18 16.1 17.1
#> 16 0.9960000000 0.0020000000 0.0010000000 0.0005000000 0.0005000000
#> 17 0.0015000000 0.9960000000 0.0015000000 0.0005000000 0.0005000000
#> 18 0.0010000000 0.0020000000 0.9960000000 0.0005000000 0.0005000000
#> 16.1 0.0003333333 0.0003333333 0.0003333333 0.9960000000 0.0030000000
#> 17.1 0.0003333333 0.0003333333 0.0003333333 0.0030000000 0.9960000000
#>
#> Model: stepwise
#> Rate: 0.003
#> Rate2: 0.001
#> Range: 0.5
#>
#> Lumpable: Not always
A simpler version of the stepwise model above, is the onestep model, in which only the immediate neighbouring integers are reachable by mutation. This model is only applicable when all alleles are integers.
mutationMatrix(model = "onestep", alleles = c("16", "17", "18"), rate = 0.04)
#> 16 17 18
#> 16 0.96 0.04 0.00
#> 17 0.02 0.96 0.02
#> 18 0.00 0.04 0.96
#>
#> Model: onestep
#> Rate: 0.04
#>
#> Lumpable: Not always