Fast Genetic Variant Call Format File Header Intelligence and Audit.
vcfheader
VCF headers contain the metadata needed to understand a dataset before inspecting variant rows. They describe file format version, reference genome, contigs, INFO and FORMAT fields, filtering definitions, samples, and other header declarations. This package extracts that information quickly and presents it in a form that is easier to inspect, validate, and share. |
TLDR
library(vcfheader)
hdr <- parse_vcf_header("file.vcf.gz")
vcfheader(
hdr,
file = "file_vcfheader.html"
)
What it does
- reads VCF headers without loading full variant records
- parses header metadata into a structured
vcf_hdrobject - extracts contigs, INFO fields, FORMAT fields, FILTER entries, ALT definitions, samples, and other header records
- performs lightweight validation and inference
- generates a portable HTML report for review and sharing
Why this is useful
The VCF header is often the fastest way to understand:
- which reference assembly was used
- what annotations are present
- how genotype fields are encoded
- what filtering or calling pipeline produced the file
- which samples are included
- whether structural variant metadata or caller-specific tags are present
Example report
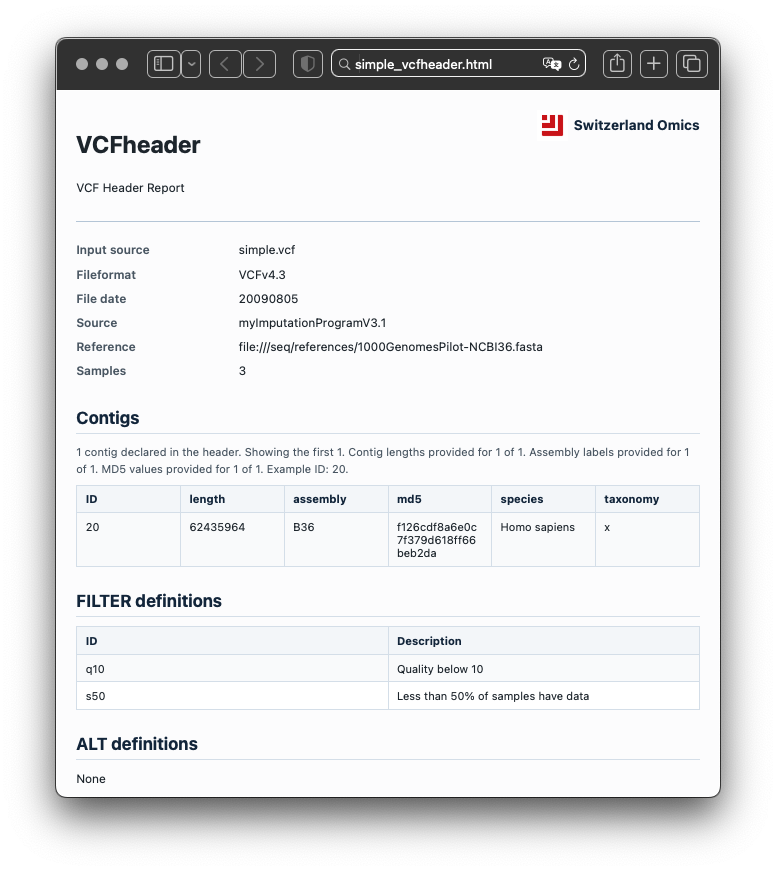
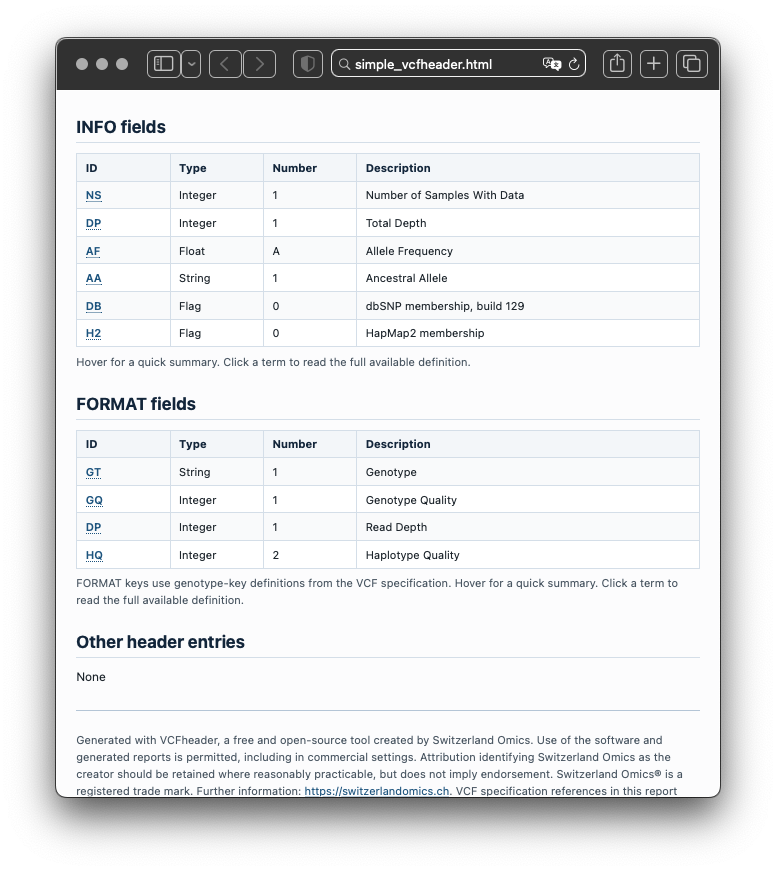
The HTML report is designed to help review dataset contents, metadata, field definitions, and filtering context in a single readable document.
Bundled examples
The package ships with small example files for offline use:
simple.vcfsv44.vcfsimple_vcfheader.html
Example usage with bundled files
library(vcfheader)
simple_vcf <- system.file("extdata", "simple.vcf", package = "vcfheader")
hdr <- parse_vcf_header(simple_vcf)
vcfheader(
hdr,
file = "simple_vcfheader.html"
)
Licence and notices
vcfheader is free and open-source software released under the GNU General Public License, version 3 (GPL-3).
Switzerland Omics is the creator of VCFheader. Attribution should be retained where reasonably practicable, but does not imply endorsement, certification, or approval of any specific use, result, or modified version.
Switzerland Omics® is a registered Swiss trade mark. No trade mark rights are granted under the software licence. Modified versions must not be presented as the original project or as endorsed by Switzerland Omics without permission.
VCF specification references may relate to samtools and the broader HTS specifications ecosystem, distributed under the MIT/Expat Licence by Genome Research Ltd. Source: Further reading: <https://www.htslib.org/doc/#file-formats.